
我认为可以从代谢和信号转导调控机制两个方面回答这个问题。

首先是巧克力,巧克力中含有咖啡因(caffeine)和可可碱(Theobromine),它俩都是甲基黄嘌呤类兴奋剂。
其主要在肠道内被吸收,由肝门静脉入肝,一部分在肝脏中被 CYP450 家族氧化代谢,转化为无活性的物质,另一部分则进入血液。
其作用于A1 和 A2A 腺苷能受体,竞争性抑制这些受体,使腺苷无法与之结合。
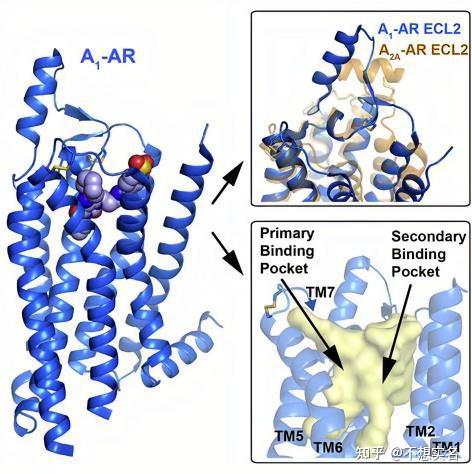
其中,A1 腺苷能受体偶联 Gi 蛋白,通过抑制腺苷酸环化酶(Adenylate cyclase,AC)降低细胞中 cAMP 含量,从而导致阳离子通道(如钙通道、钠通道)的关闭和细胞膜的超极化,导致神经系统的抑制。
好的,请记住以下这句话:咖啡因受肝脏首过效应影响,是 A1 腺苷能受体拮抗剂,可以提高神经细胞兴奋性,因此可以激活多巴胺通路。
记好了吗?我们要继续看槟榔碱(arecoline)了。
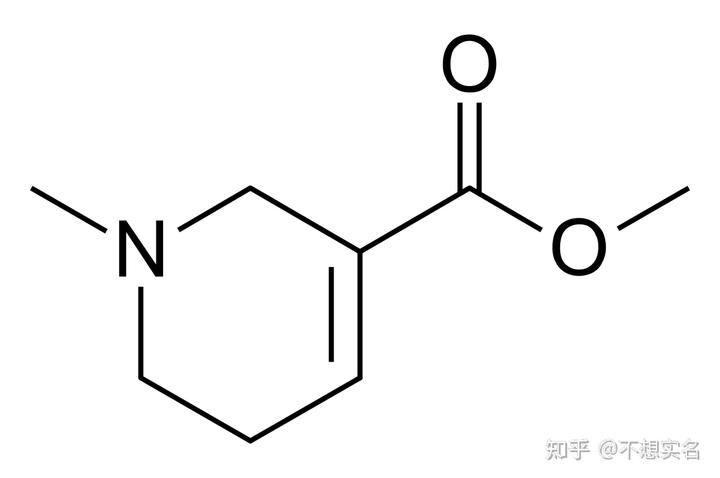
在口腔中咀嚼槟榔时,槟榔碱可以被口腔黏膜吸收,不经过肝门静脉和肝,这意味着槟榔碱不受肝脏首过效应的影响,吸收效率比咖啡因高。
其次,槟榔碱作用于毒蕈碱型乙酰胆碱受体(Muscarinic acetylcholine receptors,mAChR),尤其是 M3 型乙酰胆碱受体,并直接激活该受体,导致神经细胞兴奋与多巴胺的释放。
该受体同样为 GPCR,它与 Gq 蛋白结合,当受体激活时,Gα亚基与 Gβγ亚基解聚,从受体 C 端的 G 蛋白结合域脱离并进行信号转导,这会导致 GPCR 的 C 端暴露于细胞质内。
所以,注意到了吗,拮抗剂不会激活受体,也就不会导致受体 C 端的暴露;而激动剂相反,会导致受体 C 端的暴露,这很重要,请记住这一点。
细胞质中存在一种名为G 蛋白偶联受体激酶(G protein-coupled receptor kinase,GRK)的酶,它会磷酸化 GPCR 暴露的 C 端,这一磷酸化使受体不再能够结合 G 蛋白,从而使受体对配体的结合处于无响应状态。
随后,这个磷酸化标记还会招募β- 抑制蛋白(β-Arrestin),导致受体所在的细胞膜凹陷并形成小泡,使受体进入细胞内,不再能接触到细胞膜外的配体,这样有效的受体数量就减少了。
有效的受体数量减少了,也就意味着突触前膜释放等量神经递质引起的突触后神经元响应降低了,在乙酰胆碱的场合下,这导致了突触后神经元的兴奋性降低,从而使人萎靡不振、总感觉不对,这就是上瘾了。
因此,作为受体拮抗剂的咖啡因比作为受体激动剂的槟榔碱成瘾性弱。
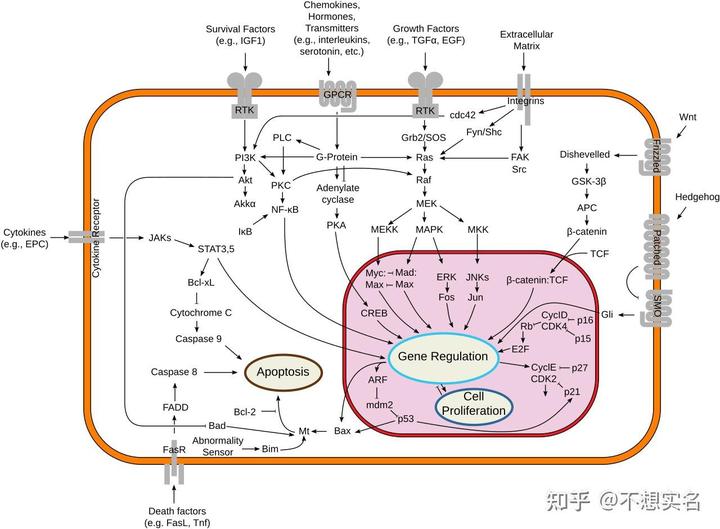
而说到危害,咖啡因最大的危害是通过拮抗 A2A 受体导致免疫系统过度激活引发炎症,以及拮抗 A1 受体导致心动过速,它们都需要极高浓度或较长时间维持高浓度的咖啡因才能达成;但 M3 胆碱能受体不一样,它被激活后会间接激活 PKC,磷酸化 Raf,通过一系列级联反应激活丝裂原活化的蛋白激酶(mitogen-activated protein kinase ,MAPK)、MEKK 和 MKK 等蛋白激酶,激活的 MAPK 等激酶转位入核,磷酸化激活 Fos、Myc、Jun 等转录因子,从而促进了细胞的增殖与向癌细胞的转化。
以上。